%matplotlib inline
Visualize Single Cell Data Using Segmentation Results
This tutorial shows how to utilize segmentation results to conduct downstream single cell analysis. The single-cell analysis part requires popularly used packages scanpy and anndata.
Here we use 10x Xenium DCIS data as an example. We mainly focus on how the segmentation results can be used for downstream single-cell analysis. The steps of model training are skipped and more details can be accessed from xxx.
Import packages & data
import sys
import random
import numpy as np
import pandas as pd
import tifffile as tiff
import matplotlib.pyplot as plt
sys.path.append('/data/aronow/Kang/spatial/Bering/cosmx')
import BeringNodeToLink_v3 as br
After the model is trained, we can use the trained model to predict the cell types and segment all spots on the whole slice. After node classification and cell segmentation is completed, we generate single cell matrix in the end.
load pretrained model
import pickle
with open('/data/aronow/Kang/spatial/Bering/demo/tutorials/plotting/models/Xenium_dcis_model_simplified.pl', 'rb') as f:
bg = pickle.load(f)
Get single cell data from whole slice
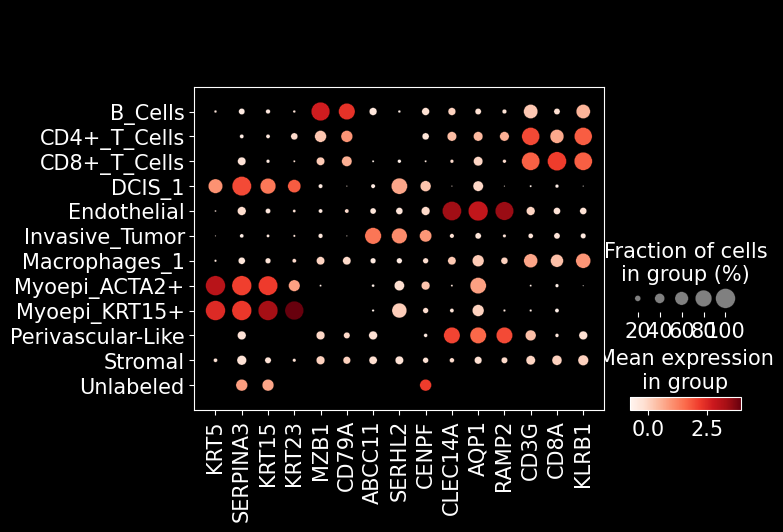
In the function br.tl.cell_annotation, we generate two single cell data, including one from direct prediction result, and the other from ensembl annotations of the prediction. More details can be found in the function description.
br.tl.node_classification(bg, bg.spots_all.copy(), n_neighbors = 30)
br.tl.cell_segmentation(bg)
df_results, adata_ensembl, adata_segmented = br.tl.cell_annotation(bg)
print(adata_segmented.shape)
print(adata_segmented.obs.head())
print(adata_ensembl.shape)
print(adata_ensembl.obs.head())
x y z features segmented labels \
molecule_id
0 2100.0261 2350.3684 19.269732 LUM 0 Stromal
1 2100.2126 2178.9346 18.475246 HAVCR2 1 Stromal
2 2100.3599 2261.9116 20.490784 RUNX1 2 Stromal
3 2100.3918 2155.2341 20.006990 TCIM 3 Stromal
4 2100.5117 2353.7695 20.749512 CXCR4 4 B_Cells
groups predicted_node_labels predicted_probability \
molecule_id
0 segmented Stromal 0.863927
1 segmented Macrophages_2 0.326667
2 segmented Stromal 0.899313
3 segmented Stromal 0.667508
4 segmented Stromal 0.667900
predicted_cells_resolution=0.01 predicted_cells \
molecule_id
0 489 489
1 320 320
2 370 370
3 313 313
4 489 489
ensembled_cells ensembled_labels
molecule_id
0 489 Stromal
1 320 Stromal
2 370 Stromal
3 313 Stromal
4 489 Stromal
(7535, 540)
n_counts n_genes cx cy
-1 4757 297 2455.268227 2433.417828
0 3396 194 2374.655610 2280.935440
1 3380 227 2371.465405 2121.776126
2 3358 250 2841.205267 2688.078428
3 3327 265 2868.166271 2333.154009
(857, 540)
n_counts n_genes cx cy predicted_labels
0 3396 194 2374.655610 2280.935440 Invasive_Tumor
1 3380 227 2371.465405 2121.776126 Invasive_Tumor
2 3358 250 2841.205267 2688.078428 Stromal
3 3327 265 2868.166271 2333.154009 Stromal
4 3219 203 2815.938887 2520.709648 Stromal
Single cell data analysis
# define function for single cell analysis from raw count matrix
import scanpy as sc
def sc_analysis(adata):
sc.pp.filter_cells(adata, min_counts = 10)
sc.pp.normalize_total(adata, target_sum = 1e3)
sc.pp.log1p(adata)
sc.pp.scale(adata) # the number of features is less than 1000, so we don't use variable genes here
sc.tl.pca(adata)
sc.pp.neighbors(adata, n_neighbors = 10)
sc.tl.umap(adata)
sc.tl.leiden(adata)
# plot Bering results and Leiden clustering
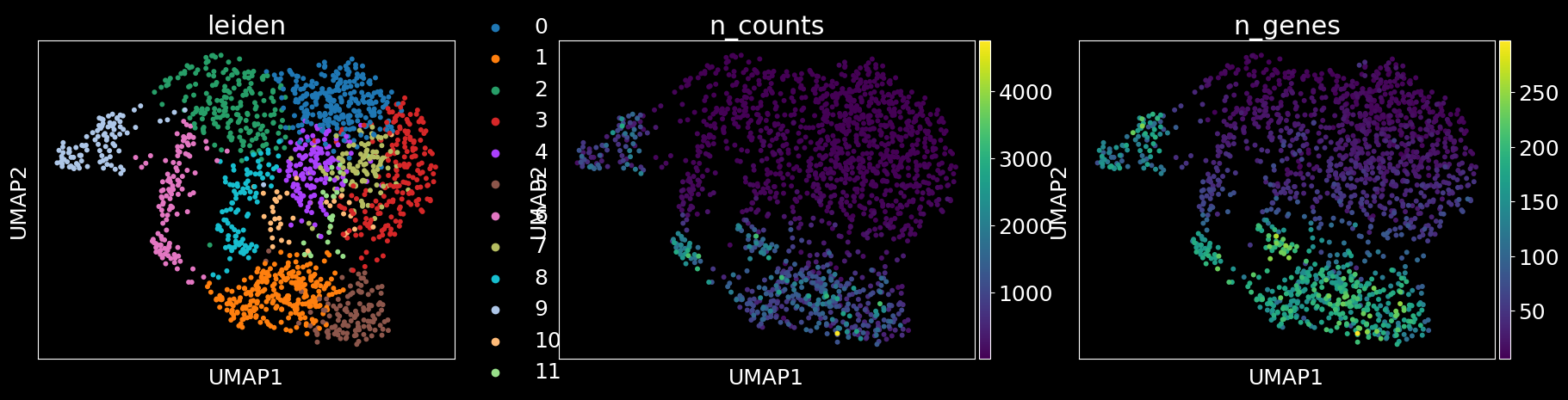
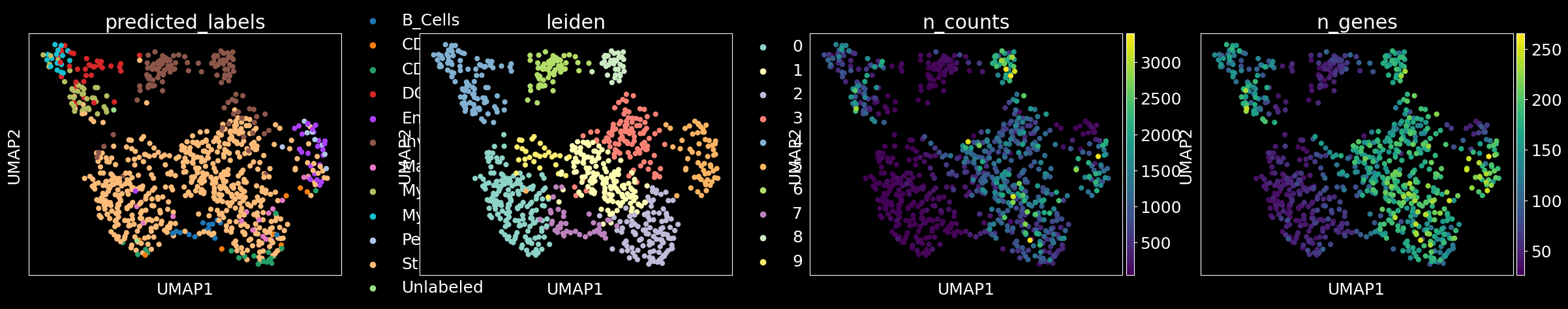
columns = ['predicted_labels','leiden','n_counts','n_genes'] if 'predicted_labels' in adata.obs.columns else ['leiden','n_counts','n_genes']
sc.pl.umap(adata, color = columns)
# analysis of segmented cells without ensembl annotations
sc_analysis(adata_segmented)
2023-07-03 17:20:19.840255: I tensorflow/core/platform/cpu_feature_guard.cc:193] This TensorFlow binary is optimized with oneAPI Deep Neural Network Library (oneDNN) to use the following CPU instructions in performance-critical operations: AVX2 AVX512F AVX512_VNNI FMA
To enable them in other operations, rebuild TensorFlow with the appropriate compiler flags.
2023-07-03 17:20:20.690205: I tensorflow/core/util/port.cc:104] oneDNN custom operations are on. You may see slightly different numerical results due to floating-point round-off errors from different computation orders. To turn them off, set the environment variable `TF_ENABLE_ONEDNN_OPTS=0`.
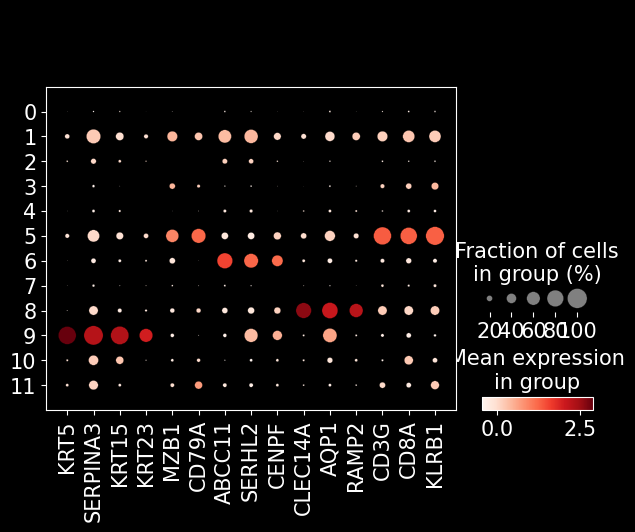
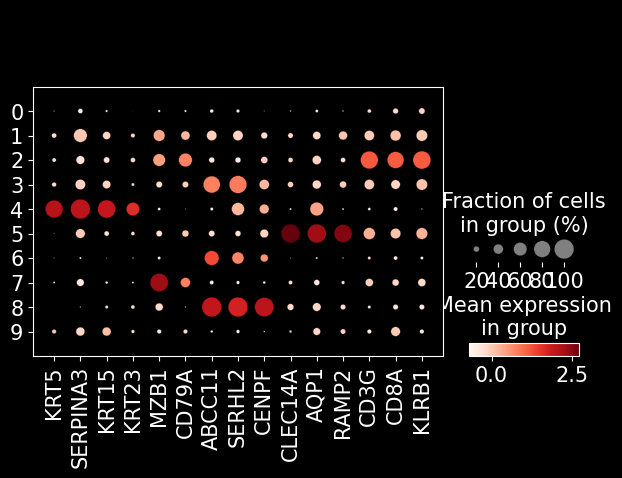
marker_genes = [
'KRT5', 'SERPINA3', 'KRT15', 'KRT23', # tumor
'MZB1', 'CD79A', # pDC
'ABCC11', 'SERHL2', 'CENPF', # epithelial
'CLEC14A', 'AQP1', 'RAMP2', # endothelial
'CD3G', 'CD8A', 'KLRB1', # T cell
]
sc.pl.dotplot(adata_segmented, marker_genes, groupby = 'leiden')
# analysis of segmented cells with ensembl annotations
sc_analysis(adata_ensembl)
... storing 'predicted_labels' as categorical
sc.pl.dotplot(adata_ensembl, marker_genes, groupby = 'leiden')
sc.pl.dotplot(adata_ensembl, marker_genes, groupby = 'predicted_labels')