%matplotlib inline
Analyze Stereoseq whole brain tissue
This tutorial shows how to apply Bering to Stereoseq whole brain data.
Stereoseq whole brain [Chen et al., 2022] data was derived from: https://db.cngb.org/stomics/mosta/. Stereoseq is a sequencing-based spatial transcriptomics technology which subcellular resolution. Here, we demonstrate the segmentation performance of Bering using E16.5 whole brain data.
Import packages & data
import random
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import Bering as br
# load data
df_spots_all = br.datasets.stereoseq_embryobrain_chen()
df_spots_seg = df_spots_all[df_spots_all['labels'] != 'background'] # foreground nodes
df_spots_unseg = df_spots_all[df_spots_all['labels'] == 'background'] # background nodes
img = None
channels = None
df_spots_seg.head()
Downloading dataset `stereoseq_embryobrain_chen` from `https://figshare.com/ndownloader/files/41409105` as `None.tsv`
| x | y | z | features | segmented | labels | region | |
|---|---|---|---|---|---|---|---|
| index | |||||||
| 1803 | 9530 | 20996 | 0 | Prox1 | 25200 | Radial glia | Pall VZ |
| 1804 | 9304 | 19163 | 0 | Prox1 | 22492 | Glutamatergic neuron | Die |
| 1806 | 9962 | 19134 | 0 | Prox1 | 29807 | Glutamatergic neuron | Die |
| 1807 | 8812 | 20901 | 0 | Prox1 | 17831 | Radial glia | Pall VZ |
| 1809 | 10028 | 18697 | 0 | Prox1 | 30784 | Glutamatergic neuron | Die |
df_spots_unseg.head() # visualize unsegmented data
| x | y | z | features | segmented | labels | region | |
|---|---|---|---|---|---|---|---|
| index | |||||||
| 14481 | 11716 | 19119 | 0 | Rgs4 | -1 | background | |
| 16791 | 8174 | 18834 | 0 | Atp1b1 | -1 | background | |
| 44115 | 11972 | 21816 | 0 | Cntnap5b | -1 | background | |
| 44122 | 9841 | 18894 | 0 | Cntnap5b | -1 | background | |
| 44492 | 11608 | 21993 | 0 | Cntnap5b | -1 | background |
# visualize the spots
x, y = df_spots_seg['x'].values, df_spots_seg['y'].values
cell_types = df_spots_seg['labels'].values
fig, ax = plt.subplots(figsize = (6, 6))
for idx, cell_type in enumerate(np.unique(cell_types)):
xc = x[np.where(cell_types == cell_type)[0]]
yc = y[np.where(cell_types == cell_type)[0]]
ax.scatter(xc, yc, s = 0.03, label = cell_type, color = np.random.rand(3,))
xb, yb = df_spots_unseg['x'].values, df_spots_unseg['y'].values
ax.scatter(xb, yb, color = '#DCDCDC', alpha = 0.2, s = 0.015, label = 'background')
h, l = ax.get_legend_handles_labels()
plt.legend(h, l, loc = 'upper right', fontsize = 8, markerscale = 15)
<matplotlib.legend.Legend at 0x2b9da9836f70>

Create Bering object
# image-dependent segmentation
bg = br.BrGraph(df_spots_seg, df_spots_unseg, img, channels)
bg
<Bering.objects.bering.Bering_Graph at 0x2b9da9955af0>
bg.segmented.head() # summary of cells
| cx | cy | cz | dx | dy | dz | d | labels | |
|---|---|---|---|---|---|---|---|---|
| segmented | ||||||||
| 0 | 9525.0 | 20996.0 | 0.0 | 20 | 9 | 0 | 20 | Radial glia |
| 1 | 9299.0 | 19172.0 | 0.0 | 39 | 37 | 0 | 39 | Glutamatergic neuron |
| 2 | 9952.0 | 19127.0 | 0.0 | 23 | 27 | 0 | 27 | Glutamatergic neuron |
| 3 | 8819.5 | 20904.5 | 0.0 | 26 | 34 | 0 | 34 | Radial glia |
| 4 | 10031.0 | 18707.0 | 0.0 | 19 | 33 | 0 | 33 | Glutamatergic neuron |
Create training data
# Build graphs for GCN training purpose
br.graphs.BuildWindowGraphs(
bg,
n_cells_perClass = 40,
window_width = 40.0,
window_height = 40.0,
n_neighbors = 20,
)
br.graphs.CreateData(
bg,
batch_size = 16,
training_ratio = 0.8,
)
Training
br.train.Training(
bg,
edge_rbf_start = 0,
edge_rbf_stop = 48,
edge_rbf_n_kernels = 64,
)
Training node classifier: 98%|█████████████████████████████████████████████████████████████████████████████████████████████████ | 49/50 [03:21<00:03, 3.12s/it]
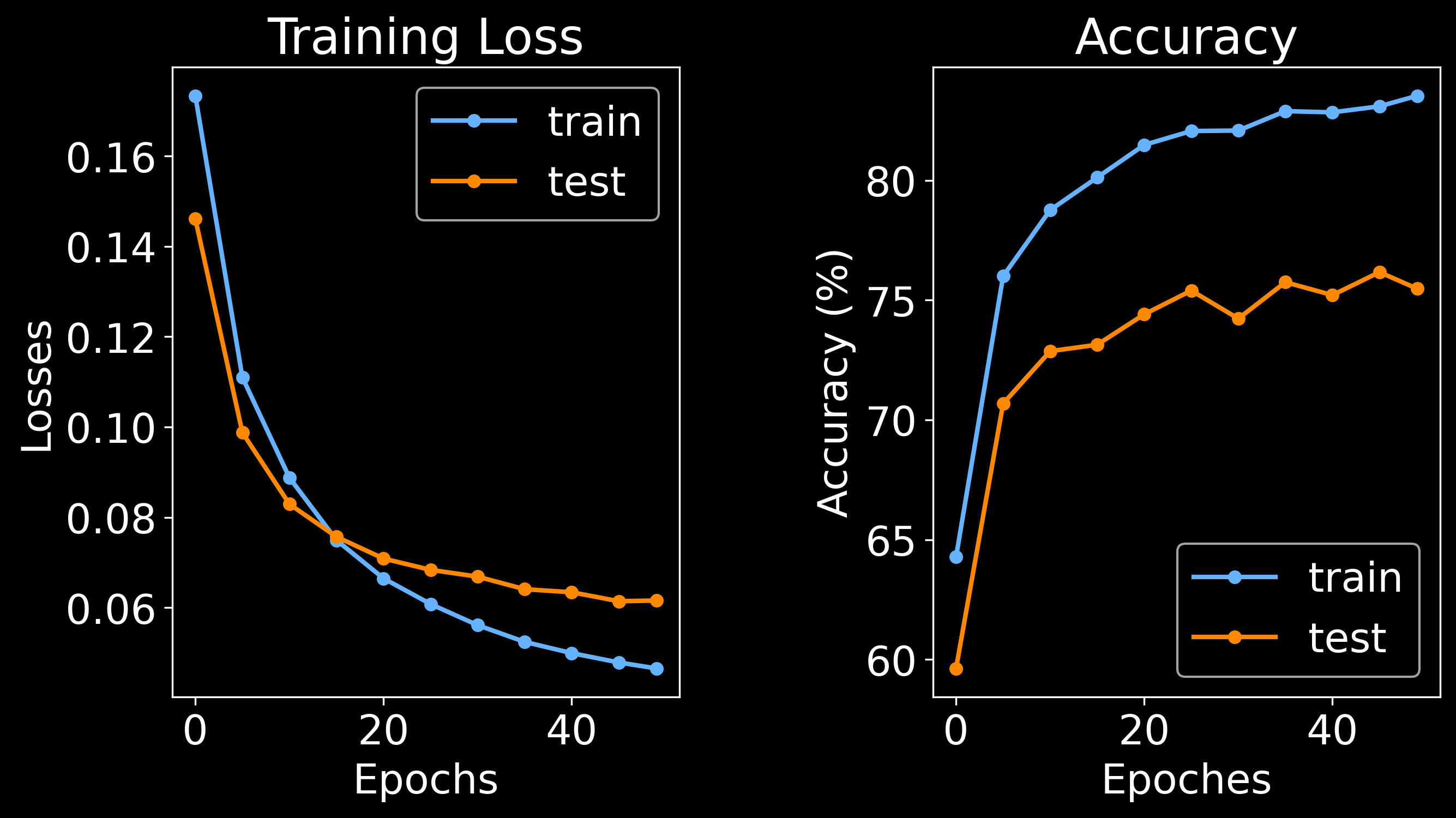
Training node classifier: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████| 50/50 [03:35<00:00, 4.31s/it]
Training edge classifier: 98%|█████████████████████████████████████████████████████████████████████████████████████████████████ | 49/50 [04:23<00:04, 4.93s/it]
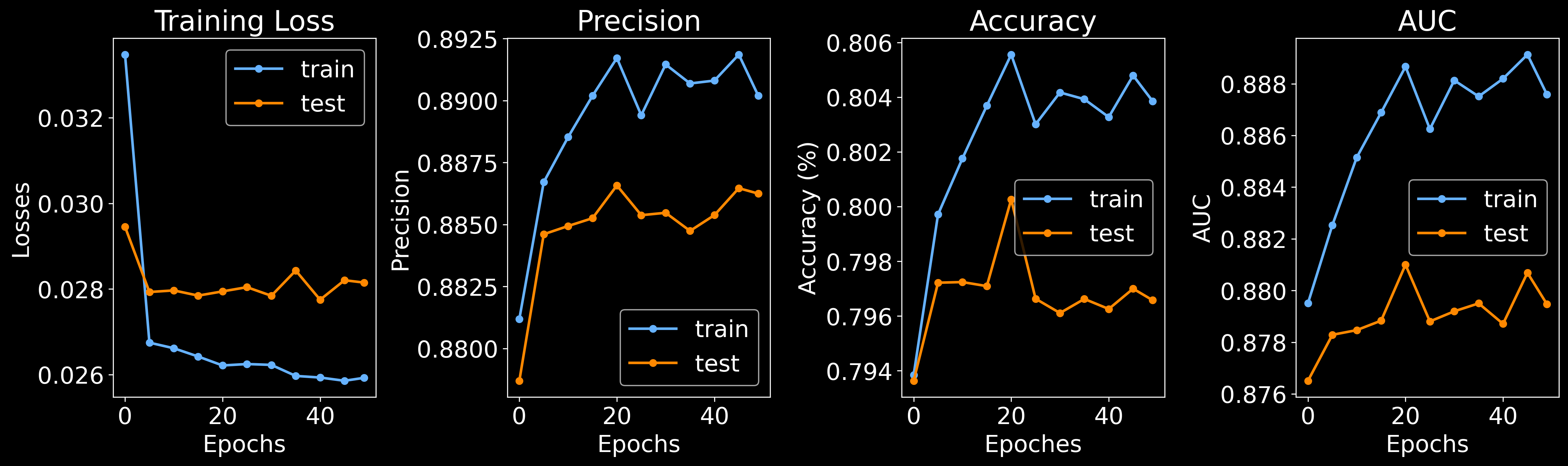
Training edge classifier: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████| 50/50 [04:33<00:00, 5.48s/it]
# save the trained model
import pickle
with open('iss_ca1_qian.pkl', 'wb') as f:
pickle.dump(bg, f)
Visualizing model
# randomly select a cell
random_cell = cells = random.sample(bg.segmented.index.values.tolist(), 1)[0]
# plot node classification
df_window_raw, df_window_pred, predictions = br.pl.Plot_Classification(
bg,
cell_name = random_cell,
n_neighbors = 10,
zoomout_scale = 8,
)

# plot cell segmentation
br.pl.Plot_Segmentation(
bg,
cell_name = random_cell,
df_window_raw = df_window_raw,
df_window_pred = df_window_pred,
predictions = predictions,
n_neighbors = 10,
zoomout_scale = 8,
use_image = True,
pos_thresh = 0.6,
resolution = 0.05,
num_edges_perSpot = 100,
min_prob_nodeclf = 0.3,
n_iters = 20,
)

After the model is trained, we can use the trained model to predict the cell types and segment all spots on the whole slice. After node classification and cell segmentation is completed, we generate single cell matrix in the end.
Node classification
Conduct node classification on the whole slice.
br.tl.node_classification(
bg, bg.spots_all.copy(),
n_neighbors = 20,
)
bg.spots_all.to_csv('spots_all.txt', sep = '\t')
Cell segmentation
br.tl.cell_segmentation(bg)
Get single cells
df_results, adata_ensembl, adata_segmented = br.tl.cell_annotation(bg)
df_results.to_csv('results.txt', sep = '\t')
adata_ensembl.write('results_cells_ensembled.h5ad')
adata_segmented.write('results_cells_segmented.h5ad')
print(f'Ensembled anndata: {adata_ensembl.shape}')
print(f'Segmented anndata: {adata_segmented.shape}')
... storing 'predicted_labels' as categorical
Ensembled anndata: (1642, 223)
Segmented anndata: (7918, 223)
Single cell data analysis
import scanpy as sc
sc.settings.set_figure_params(dpi=80)
# run standard analysis on the ensembled anndata
adata_ensembl = br.tl.cell_analyze(adata_ensembl, min_counts = 5)
# first check the data quality of segmented cells
sc.pl.umap(adata_ensembl, color = ['n_counts','n_genes'])
2023-08-22 17:52:36.256254: I tensorflow/core/platform/cpu_feature_guard.cc:193] This TensorFlow binary is optimized with oneAPI Deep Neural Network Library (oneDNN) to use the following CPU instructions in performance-critical operations: AVX2 AVX512F AVX512_VNNI FMA
To enable them in other operations, rebuild TensorFlow with the appropriate compiler flags.
2023-08-22 17:52:37.332336: I tensorflow/core/util/port.cc:104] oneDNN custom operations are on. You may see slightly different numerical results due to floating-point round-off errors from different computation orders. To turn them off, set the environment variable `TF_ENABLE_ONEDNN_OPTS=0`.

# plot leiden clustering and predicted labels from Bering
fig, axes = plt.subplots(1, 2, figsize = (15, 5))
axes[0] = sc.pl.umap(adata_ensembl, color = ['leiden'], ax = axes[0], show = False)
axes[1] = sc.pl.umap(adata_ensembl, color = ['predicted_labels'], ax = axes[1], show = False)
plt.subplots_adjust(wspace = 0.5)
plt.tight_layout()
plt.show()
